Scientists on Thursday unveiled the most exhaustive database yet of the proteins that form the building blocks of life, in a breakthrough observers said would "fundamentally change biological research".
Every cell in every living organism is triggered to perform its function by proteins that deliver constant instructions to maintain health and ward off infection.
Unlike the genome – the complete sequence of human genes that encode cellular life – the human proteome is constantly changing in response to genetic instructions and environmental stimuli.
Understanding how proteins operate – the shape in which they end up, or "fold" into – within cells has fascinated scientists for decades.
But determining each protein's precise function through direct experimentation is painstaking.
Fifty years of research have until now yielded only 17 percent of the human proteome's amino acids, the subunits of proteins.
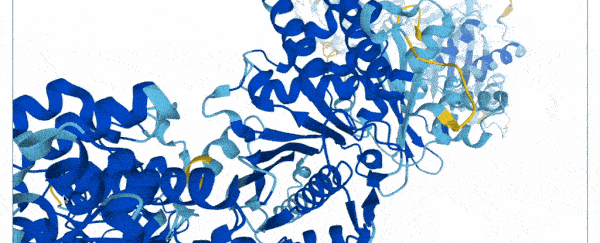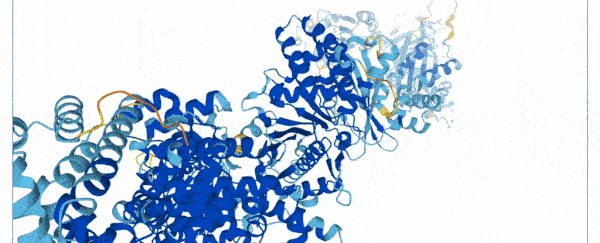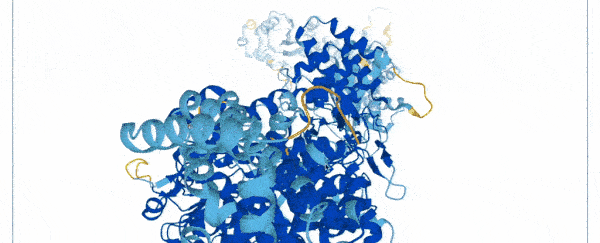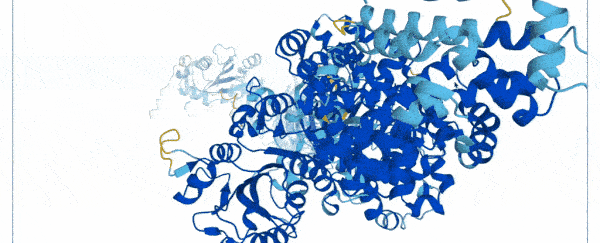
On Thursday, researchers at Google's DeepMind and the European Molecular Biology Laboratory (EMBL) unveiled a database of 20,000 proteins expressed by the human genome, freely and openly available online.
They also included more than 350,000 proteins from 20 organisms such as bacteria, yeast, and mice that scientists rely on for research.
To create the database, scientists used a state-of-the-art machine learning program that was able to accurately predict the shape of proteins based on their amino acid sequences.
Instead of spending months using multi-million dollar equipment, they trained their AlphaFold system on a database of 170,000 known protein structures.
The AI then used an algorithm to make accurate predictions of the shape of 58 percent of all proteins within the human proteome.
This more than doubled the number of high-accuracy human protein structures that researchers had identified during 50 years of direct experimentation, essentially overnight.
The potential applications are enormous, from researching genetic diseases and combating anti-microbial resistance to engineering more drought-resistant crops.
'Protein-folding problem'
Paul Nurse, winner of the 2001 Nobel Prize for Medicine and director of the Francis Crick Institute, said Thursday's release was "a great leap for biological innovation".
"With this resource freely and openly available, the scientific community will be able to draw on collective knowledge to accelerate discovery, ushering in a new era for AI-enabled biology," he said.
John McGeehan, director for the Centre for Enzyme Innovation at the University of Portsmouth, whose team is developing enzymes capable of consuming single-use plastic waste, said AlphaFold had revolutionized the field.
"What took us months and years to do, AlphaFold was able to do in a weekend. I feel like we have just jumped at least a year ahead of where we were yesterday," he said.
The ability to predict a protein's shape from its amino acid sequence using a computer rather than experimentation is already helping scientists in a number of research fields.
AlphaFold is already being used in research into cures for diseases that disproportionately affect poorer countries.
One US-based team is using the AI prediction to study ways of overcoming strains of drug-resistant bacteria.
Another group is using the database to better understand how SARS-CoV-2, the virus that causes COVID-19, bonds with human cells.
Venki Ramakrishnan, winner of the 2009 Nobel Prize for Chemistry, said Thursday's research, published in the journal Nature, was a "stunning advance" in biological research.
He said AlphaFold had essentially solved the so-called "protein-folding problem", which argued that the 3D structure of a given protein should be determinable from its amino acid sequence, and which had puzzled scientists for half a century.
Given that the number of shapes a protein could theoretically take is astronomically large, the protein-fold problem was partly one of processing power.
The task was so daunting that in 1969 US molecular biologist Cyril Levinthal famously theorized that it would take longer than the age of the known universe to enumerate all possible protein configurations using brute calculation.
But with AlphaFold capable of performing a mind-dizzying number of calculations every second, the problem stood no chance when faced with AI and algorithms.
"It has occurred long before many people in the field would have predicted," Ramakrishnan said.
"It will be exciting to see the many ways in which it will fundamentally change biological research."